WGCNA
WGCNA分析基于两个假设:1.相似表达模式的基因可能存在共调控、功能相关或处于同一通路,2.基因网络符合无尺度分布。基于这两点,可以将基因网络根据表达相似性划分为不同的模块进而找出枢纽基因。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
| #0.准备工作---- 需要准备的包
rm(list = ls()) #一键清空~
options(stringsAsFactors = F)
library(stringr)
library(limma)
library(ggplot2)
library(dplyr)
library(WGCNA)
library(data.table)
library(tidyverse)
library(openxlsx)
#导入数据
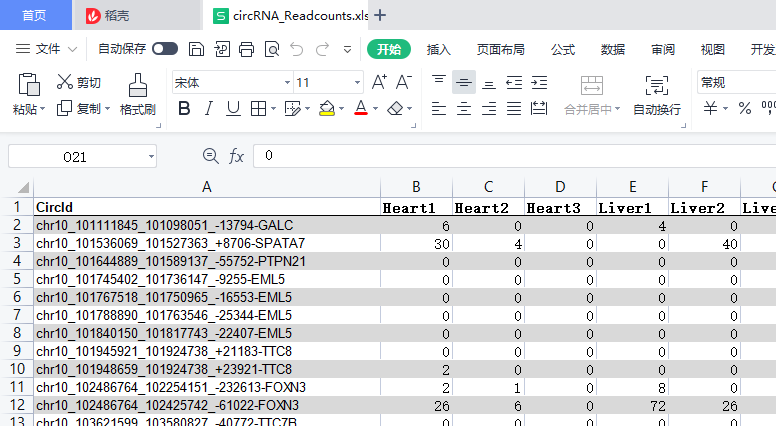
exp = read.xlsx("circRNA_Readcounts.xlsx",sheet =1)
row.names(exp) = exp[ ,1]
exp = exp[ ,-1]
|
由于我的circRNA预测出的过多,导致大量存在0,1,2等个位数的counts,处理
1
2
3
4
5
6
7
8
9
| 录入数据预处理,过滤
keep <- rowSums(exp>0) >= floor(0.75*ncol(exp)) #floor 向下取整,也就是一行所有数字加起来的0.75倍,如果有,就存留
table(keep)
filter_count <- exp[keep,]
filter_count[1:21,1:21]
dim(filter_count)
exp = filter_count
View(head(exp))
#处理完毕后过滤掉大量低counts的基因,非编码数目从6w降低到2w左右
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
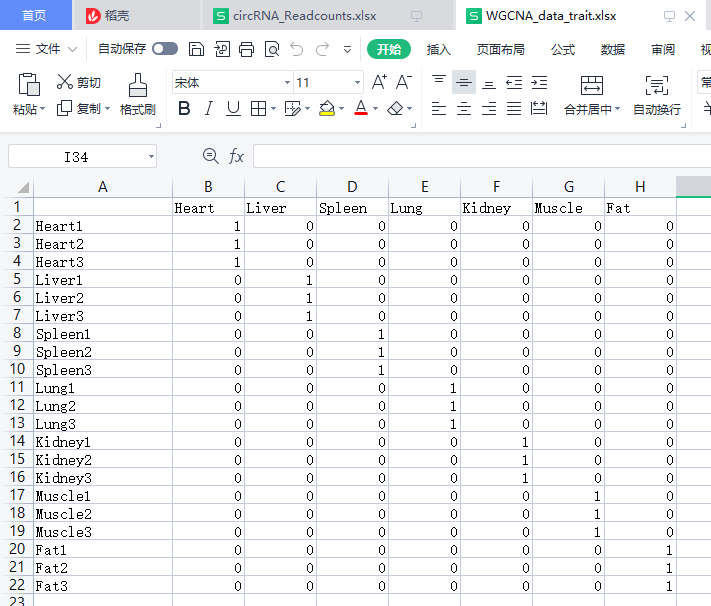
| #录入表型信息
pd <- read.xlsx("WGCNA_data_trait.xlsx",sheet = 1)
row.names(pd) = pd[ ,1]
pd = pd[ ,-1]
pd <- data.frame(pd)
head(pd)
exp = exp[,colnames(exp) %in% rownames(pd)]
p = identical(rownames(pd),colnames(exp));p
######标准化
#将所有数据进行标准化
library(limma)
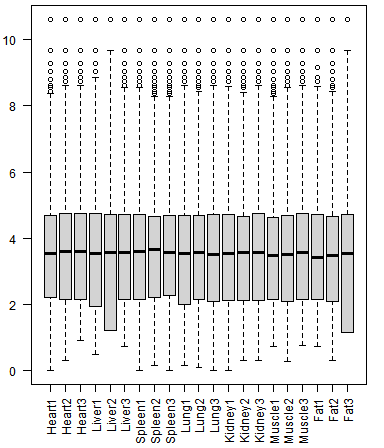
png(filename = "1-raw-sample-stat-boxplot.png")
par(mai = c(1.3,1,0.1,1) )
boxplot(exp,las=2)
dev.off()
exp=log2(exp+1)
exp=normalizeBetweenArrays(exp)
png(filename = "2-normal-sample-stat-boxplot.png")
par(mai = c(1.3,1,0.1,1) )
boxplot(exp,las=2)
dev.off()
|